
US FDA Warning Letters

ExThera Medical is facing legal trouble after two patients died following treatment with its blood filtration device. The company's former chief regulatory officer has pled guilty to failing to report adverse events, while ExThera signed a deferred prosecution alert and agreed to pay $6.45m.

The US FDA is warning that anti-choking devices can prove ineffective in emergencies and that methods established by the American Red Cross and the American Heart Association to remove blockages from choking victims should be tried first.

In two warning letters posted Feb. 24, the US FDA cited a device manufacturer with clinical trial violations and another for failing to meet current good manufacturing practices.

Twenty-one companies received US FDA warning letters in December and January. About half were part of a crackdown on unregistered vendors of breast binders, while other recipients included Abbott and Magellan Diagnostics.

A warning letter from the US FDA citing concerns of some Abbott continuous glucose monitors will not stop the company from launching a novel diabetes sensor later this year as planned.

The US FDA has warned that Magellan Diagnostics blood lead tests may provide inaccurately high results, particularly with tubes from ASP Global. Magellan’s tests had previously been recalled over inaccurate low results. The FDA also issued warning letters to both firms.
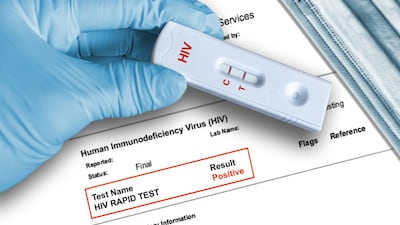
The US FDA has sent warning letters to four companies that it says distribute HIV sample collection kits for home use without proper regulatory authorization.

Last year was a busy one for the medtech industry, with major advances in technology as well as big changes at regulatory agencies. Get a peek at the stories our readers couldn't miss.

In a letter to its customers, Medtronic said it had also received 41 complaints indicating that the catheters were not retaining their intended shape when bent. The FDA issued an early alert about the recall in August but did not at that time assign a recall class.

While the US Department of Health and Human Services presented its Dec. 18 actions limiting access to gender-affirming care as necessary to protect children, a dozen warning letters issued to manufacturers and vendors of chest binders focus on lack of regulatory clearance, rather than consumer age.

The US FDA issued eight warning letters in September and October, with ultrasound, surgical and mobile apps among the affected product types.

US FDA inspections of three Philips manufacturing sites earlier this year resulted in a September warning letter that claimed the company was not in conformity with current good manufacturing practices. Philips says it is addressing the agency’s concerns and working to enhance its quality systems.

Two recent warning letters from the US FDA provide the companies with 30 business days to respond instead of the usual 15. Though not a regulatory requirement, the 15-day time frame has become the standard.

Dexcom has endured recalls, layoffs and leadership change, but interim CEO Jake Leach said the company remains committed to innovating next-gen devices.

The US FDA issued two safety alerts concerning associated risks from unauthorized medical devices. The alerts are consistent with a larger effort to clamp down on consumer goods that have not been subject to agency review.

Legal experts say a recent warning letter from the US FDA to a maker of dental products could upend a long-standing dental industry view that its operations and products are exempt from agency oversight.

The US FDA posted nine device-related warning letters in August, covering clinical research labs, laser therapy, orthopedics and more.

The US FDA posted three warning letters in July, covering medical supply kits, wearables and orthopedics.

The reason for the correction is that if the device momentarily activates but does not cut or staple tissue, providers can be inadvertently locked out during surgical procedures. When a lockout occurs, additional steps are required to open the device and remove it from tissue.

A panel of regulatory pros offered stakeholders tips for avoiding FDA citations after facility inspections. The July 29 webinar, hosted by ProPharma and Hyman Phelps and McNamara, follows the agency’s May announcement that it plans to up random foreign inspections.