
Post-Marketing Regulation & Studies

Pink Sheet editors discuss the impact of a federal judge’s decision that the recent ACIP membership turnover and vaccine schedule changes likely violated statutes, as well as the FDA’s look at new opioid disposal requirements.

After mandating mail back envelopes be available, the FDA now is considering whether disposal options that would allow opioids to be thrown in the trash also should be required, potentially adding to the expense for sponsors.

The FDA's widely referenced scale-up and post-approval changes (SUPAC) guidelines could be modernized to acknowledge evolving science and conflicts with newer recommendations.
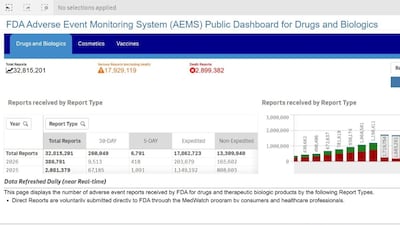
The US FDA's new AEMS database included adverse event reports for drugs and vaccines on March 11, while device, human food and tobacco product data will be added in May.

Just a month after sending Novo Nordisk a warning over an ad for its Wegovy pill, the US FDA has sent another warning raising concerns over the firm’s safety reporting compliance.

The European Medicines Agency has explained how companies can implement ICH E2D(R1), a revised guidance on post-approval safety data that aims to clarify how to manage safety data derived from solicited sources such as social media, market research programs, and patient support programs.

Results from two studies did not establish efficacy or a causal link to serious adverse events in both subgroups, but the Comirnaty labeling update is notable given the sea change in US vaccine regulation and policy since Pfizer submitted the sBLA in April 2025.

The idea proposed by industry could be an attempt to increase first-cycle approvals and reduce overall review times, but the FDA has questions.

Amgen's refusal to voluntarily withdraw the rare disease treatment could force the FDA to employ its formal process to remove it from the market.
Two flu vaccine labels already include febrile seizure in the clinical trial and postmarketing experience sections of the label, but the FDA wants all of them to add a warning of the adverse event.

The US FDA’s comprehensive review found no increased risk of suicidal ideation and behavior with the use of GLP-1s to treat obesity, leading the agency to request existing warnings based on older drugs be removed from labeling.

The FDA outlined its plans for “Sentinel 3.0,” including a data hub that would speed queries, during PDUFA VIII negotiations as industry questions whether user fee revenue should continue funding the program.

Medical device sponsors can use real-world evidence without submitting identifiable patient-level data under a new guidance that is expected to expand to drugs and biologics.
As differences emerged between FDA staff and senior political leaders over its COVID-19 safety review, CBER Director Vinay Prasad added an old colleague and critic of US COVID-19 policy to the center.

The FDA included storage and handling instructions common in labeling in its approval announcement, suggesting concerns remain about the potential for NDMA to form after products are distributed.

Incentives are “probably the only solution” to encouraging companies to optimize cancer drugs, but this will require funding and systemic changes, the chair of the European Medicines Agency’s oncology working party says.

After 10 years of adding suffixes to all new biologic and biosimilar nonproprietary names, FDA officials are considering whether it is still necessary for pharmacovigilance purposes.
After two deaths tied to the gene therapy, Sarepta and the FDA agreed to new labeling for Elevidys, adding a black box warning about liver injury along with suggested liver and cardiac monitoring.

Real-world evidence (RWE) studies in India face challenges like lack of data reliability and uniformity and absence of clear guidelines but firms like Bharat Serums have scored a regulatory win. Pink Sheet examines the RWE landscape and the BSV case study for lessons in beating the odds

The approval is more limited than hoped, but GSK said further studies will help make Blenrep a major treatment in multiple myeloma.