
Product Reviews
Approvals

The European Medicines Agency is set to adopt opinions on whether marketing approval should be granted to five new products, including three orphans. A decision is also due on a previously-rejected Alzheimer’s drug that was under re-examination.

The agency is conducting further investigation of reports of altered skin sensation that occurred more frequently with the 7.2 mg semaglutide dose than with lower doses of the GLP-1 inhibitor.

Smaller biotech companies without the regulatory resources of big pharma should approach the UK medicines regulator and health technology appraisal body for early, informal discussions on how to generate the right evidence.

Asieris Pharmaceuticals' hexaminolevulinate, for treating high-grade squamous intraepithelial lesions, is among the latest products that have been filed for review by the European Medicines Agency for potential EU marketing approval.
Complete Response Letters

An HHS administrative law judge will preside at the formal evidentiary hearing, which stems from a 2019 complete response letter that was followed by administrative and legal appeals, even though Vanda said it would only accept the FDA commissioner as presiding officer.

The FDA’s contrasting decisions on leucovorin and idebenone reveal a tightening regulatory stance where only therapies showing strong mechanistic rationale and large, credible treatment effects can overcome the limitations of nontraditional evidence sources.

The interim facility inspection designation is being used as the basis for complete response letters and is part of a pattern of agency regulatory and administrative measures to avoid missing user fee deadlines, experts told the Pink Sheet.

Moderna’s mRNA-1010 is an outlier among RTF recipients, which are dominated by rare disease and neuroscience candidates, such as Axsome’s fibromyalgia drug and Neuvivo’s ALS immunotherapy.
Drug Review Profiles

Single-item questionnaires were straightforward for patients to understand and measured clinically meaningful outcomes, while multi-symptom assessments lacked content validity, the FDA said in its review of Vanda’s Nereus.

The Pink Sheet’s Drug Review Profile examines the clinical development and US FDA review timeline for Vanda’s tradipitant to prevent motion-induced vomiting.

Tradipitant labeling states that safety for prevention of motion-induced vomiting has not been established for more than 90 total doses, reflecting FDA pharm/tox reviewer concerns about the lack of a chronic toxicity study in a nonrodent animal species.

Saol Therapeutics cannot conduct a new clinical trial of SL1009 in ultra-rare mitochondrial disorder, but aims to answer the FDA's complete response letter with new looks at available data.
EU CHMP
The European Medicines Agency is set to adopt opinions on whether marketing approval should be granted to five new products, including three orphans. A decision is also due on a previously-rejected Alzheimer’s drug that was under re-examination.
Asieris Pharmaceuticals' hexaminolevulinate, for treating high-grade squamous intraepithelial lesions, is among the latest products that have been filed for review by the European Medicines Agency for potential EU marketing approval.

Novartis’s ianalumab, for treating Sjögren’s disease, is also among the new drugs that the European Medicines Agency has started reviewing for potential pan-EU marketing approval.

This is an update of recommendations from the European Medicines Agency's Committee for Medicinal Products for Human Use on the authorization of new medicines in the EU, and updates on EU marketing authorization changes recommended by the CHMP.
Post-Marketing Regulation & Studies

Pink Sheet editors discuss the impact of a federal judge’s decision that the recent ACIP membership turnover and vaccine schedule changes likely violated statutes, as well as the FDA’s look at new opioid disposal requirements.

After mandating mail back envelopes be available, the FDA now is considering whether disposal options that would allow opioids to be thrown in the trash also should be required, potentially adding to the expense for sponsors.

The FDA's widely referenced scale-up and post-approval changes (SUPAC) guidelines could be modernized to acknowledge evolving science and conflicts with newer recommendations.
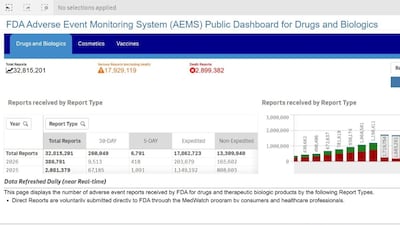
The US FDA's new AEMS database included adverse event reports for drugs and vaccines on March 11, while device, human food and tobacco product data will be added in May.
US Advisory Committees

With the US CDC Advisory Committee on Immunization Practices sidelined by the courts, the Pink Sheet imagined how a past version of the panel may have spent the spring, including tracking a fast‑moving pipeline of flu, RSV, Lyme, dengue, and next‑generation combination vaccines.
Vaccine manufacturers warned the FDA vaccines advisory committee that international genetic resource rules are delaying influenza virus sharing and creating regulatory hurdles that could undermine seasonal vaccine preparedness.

Trivalent formulations for the 2026-2027 northern hemisphere flu season address the swift rise of subclade K, but the persistent need for broader antigen coverage keeps quadrivalent possibilities in mind.

Significant viral drift and a temporarily rebuilt committee set the stage for a high‑stakes VRBPAC vote on updates to the three influenza vaccine strains for the 2026–2027 season.